Direct Force Field 8
Direct Force Field (DFF) is a specialized software that makes and uses high quality force field parameters with unlimited coverage to meet the ever-increasing demands in atomistic simulations.
The latest version is DFF 8, which has the following major components:
Graphic User Interface
The GUI provides visualization and editing of molecular models. It supports most functionalities of DFF package, as well as interfaces to external software: Gaussian, GROMACS and LAMMPS.
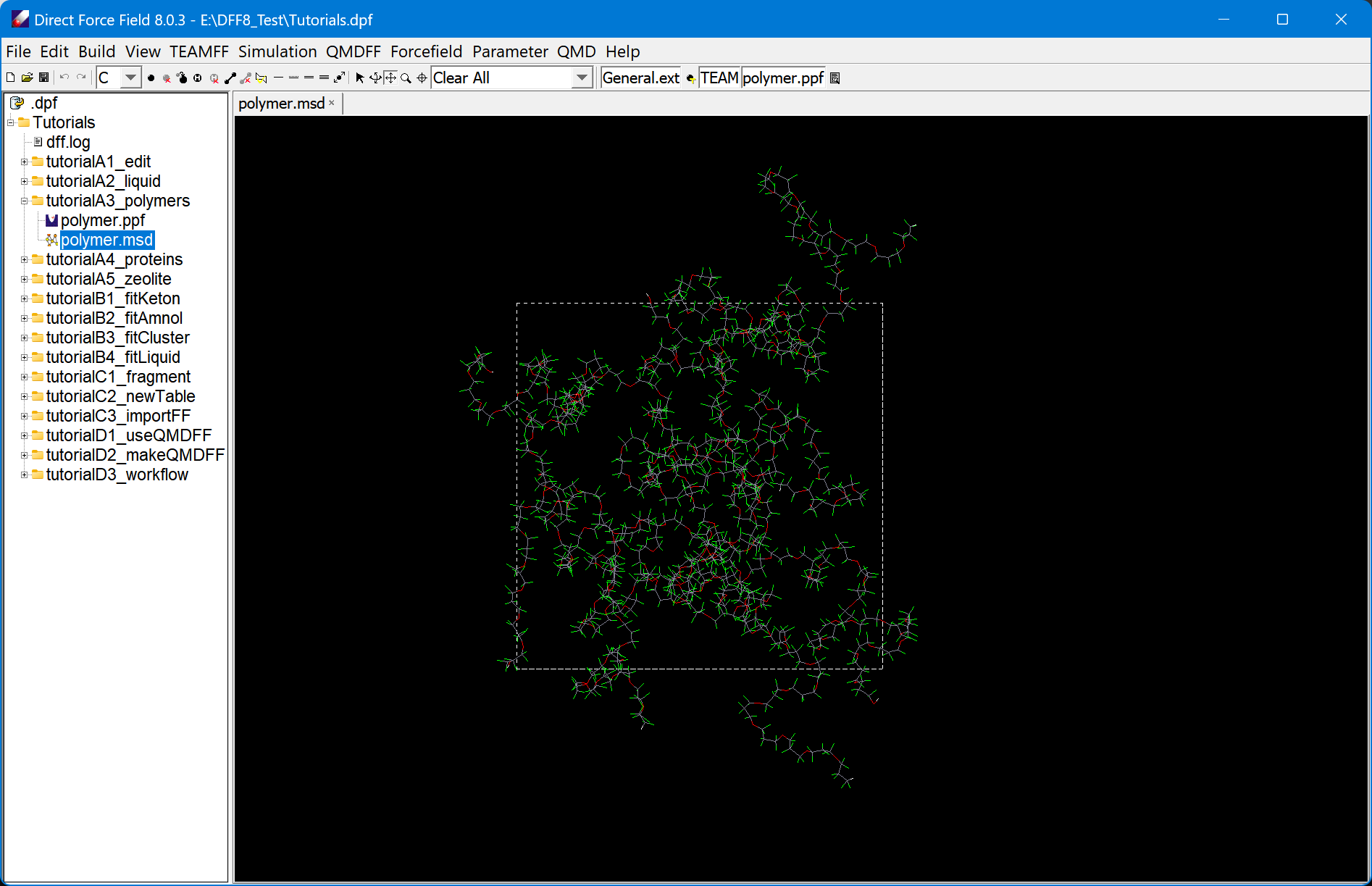
Build Training Set and Generate Data
DFF 8 has automatic data generator that automatically collect the required ab initio data for parametrization. For any target molecule and force field type, it automatically make molecular fragments, prepare quantum mechanical computations for each of the fragments to sample its potential energy surfaces, and prepare the quantum mechanics data (QMD) for parameterization.
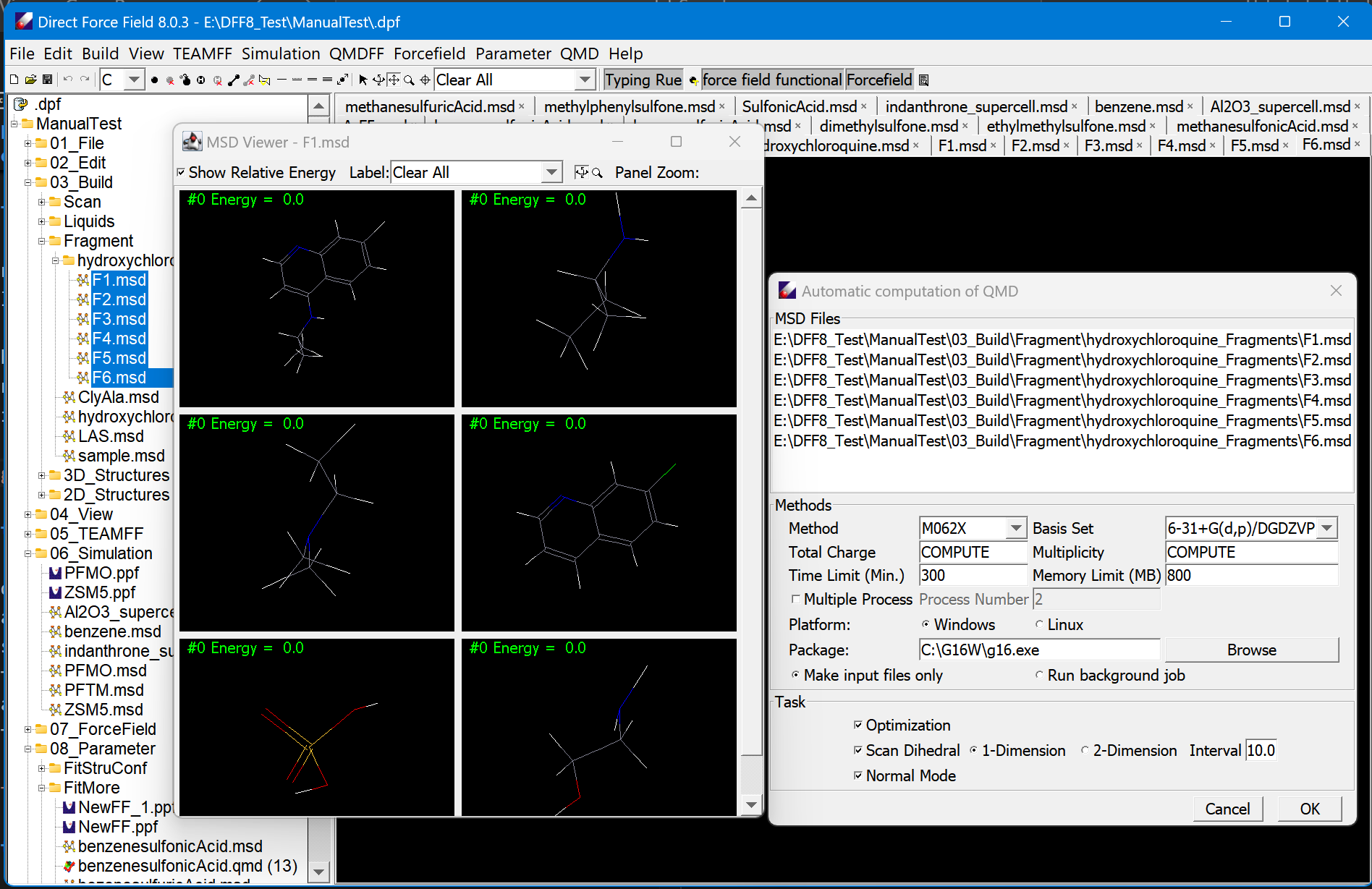
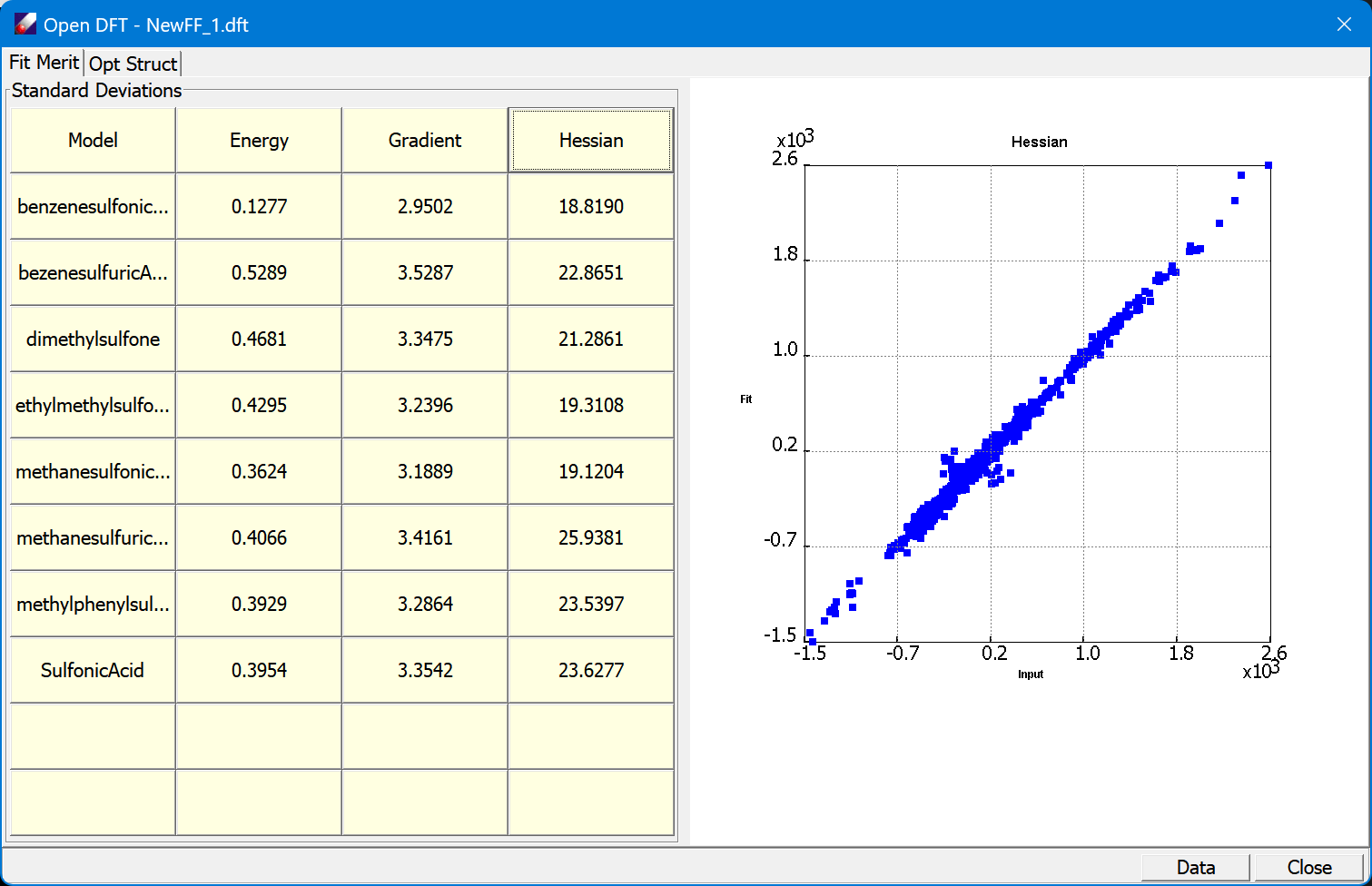
Parametrization
DFF includes all essential tools for making high quality force field parameters. It derives electrostatic and valence parameters by fitting QMD with default Lennard-Jones parameters that are taken from the existing research results. With the electrostatic and valence parameters are obtained, the default LJ parameters can be optimized by fitting liquid data.
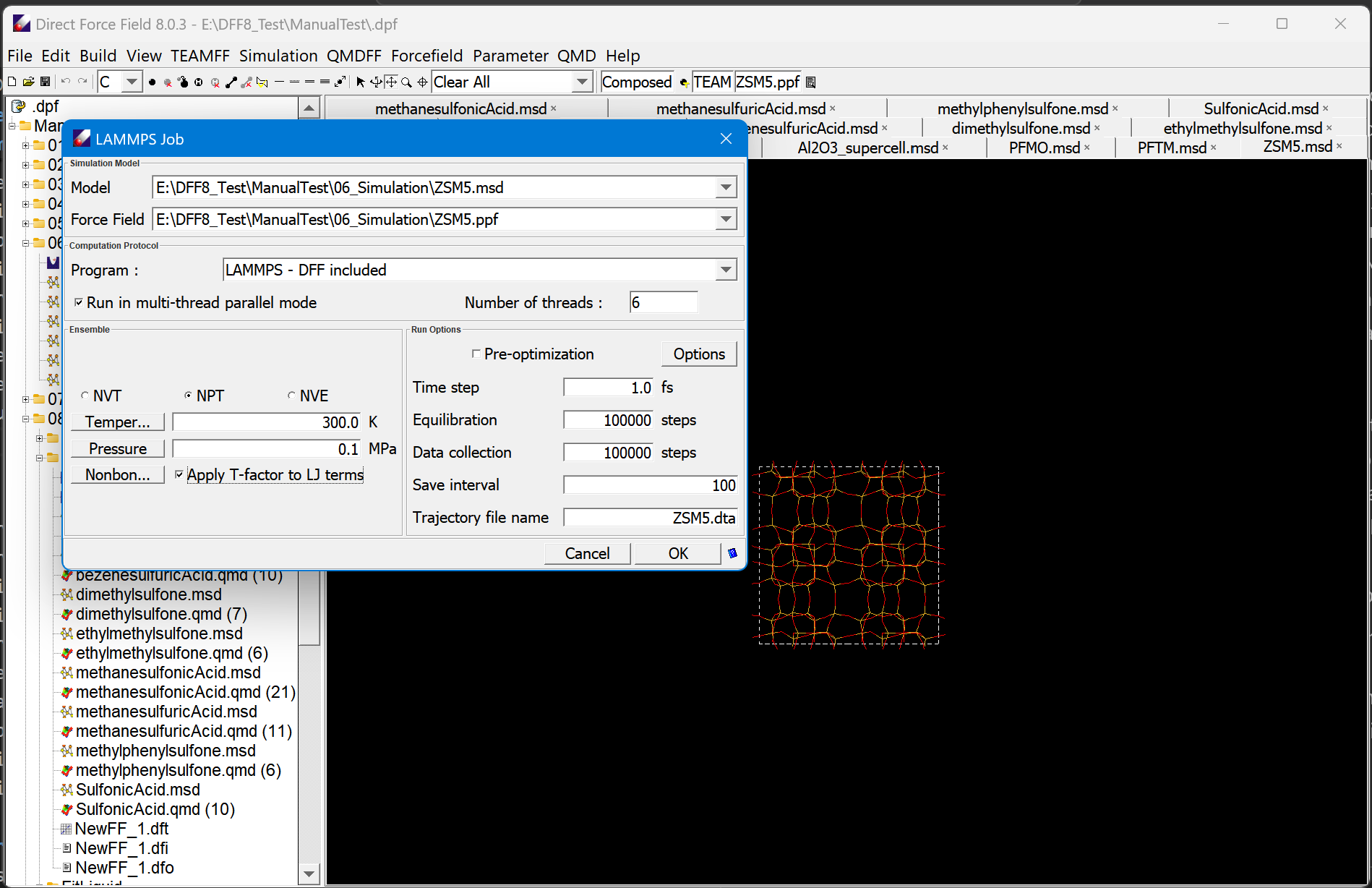
Simulation
A native simulation engine is implemented for quick validation of the force field parameters. energy minimization and molecular dynamics. Two different algorithms, the conjugate gradient and variable metric methods, are implemented for energy minimization. Restrained energy minimization can be performed to explore energy profiles. In the molecular dynamics, the Verlet velocity algorithm is implemented. NVT, MVE and NPT ensembles can be simulated. This program is useful for validating parameters, optimizing VDW parameterization and performing basic molecular dynamics simulations.
Force Field Integrator (FFI) is an import product to support other simulation engines. It is implemented to enable a seamless integration of the same kind of force field parameters from different sources. For example, you can integrate parameters for proteins from the AMBER or CHARMM force field with parameters for ligands from the TEAM databases. The program validates the integration, ensures the result parameters can be used directly by the simulation engines. It supports several popular simulation engines such as GROMACS, AMBER, CHARMM, and DISCOVER by exporting required topologic and parameter files. In addition, it includes a user interfaces to support molecular dynamics simulation engine GROMACS directly.
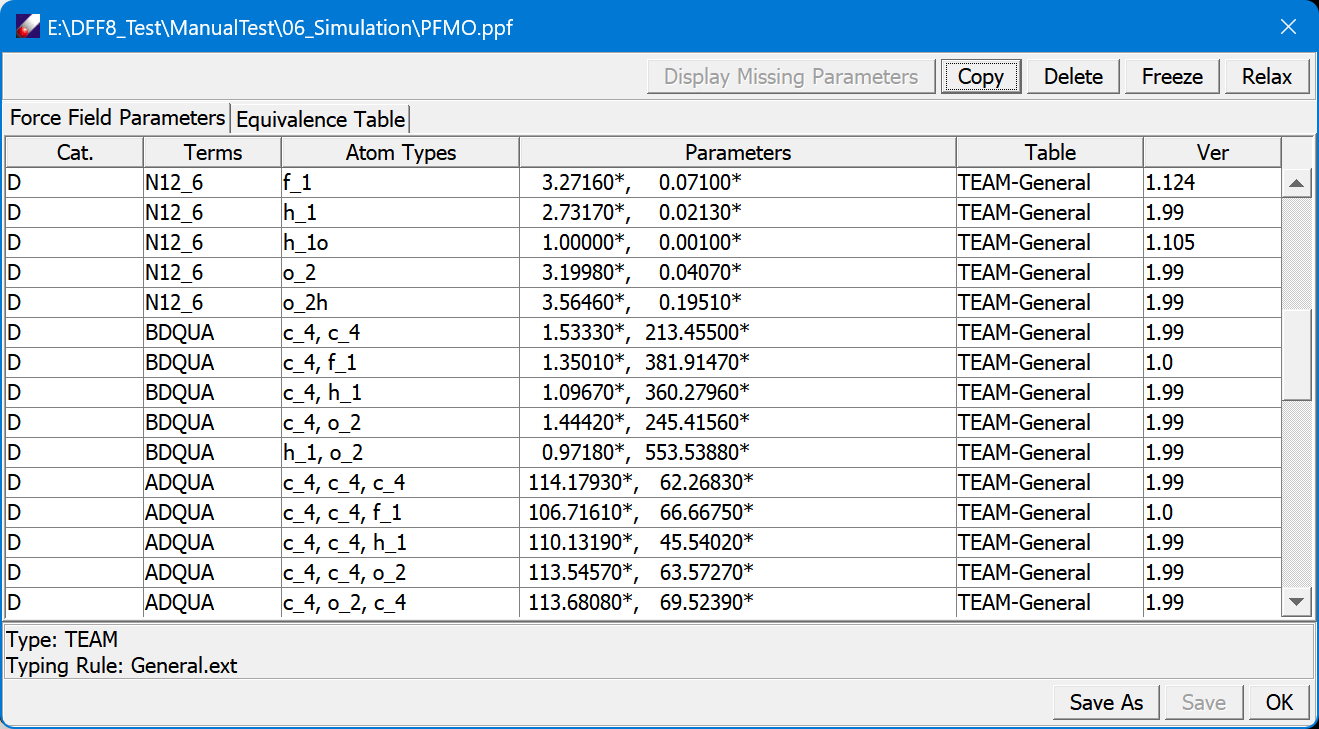
Force Field Managements
DBM is a database manager for making and managing custom TEAM databases. For maintaining a standard of supporting all customers, the default TEAM force field databases are not extensible by the end users. In order to extend the coverage of the default databases, you need to make your own databases. The customer-made TEAM databases can be used standalone or together with other TEAM databases as long as they are in the same force field type (e.g. AMBER, CHARMM, or CFF). The database manager controls how the data flows, validates the data, and indexes the data entries for fast access. The following data can be stored in TEAM databases �C quantum chemistry data, molecular fragment structure data, force field parameters, parameterization validation results, and any user-input text notes.
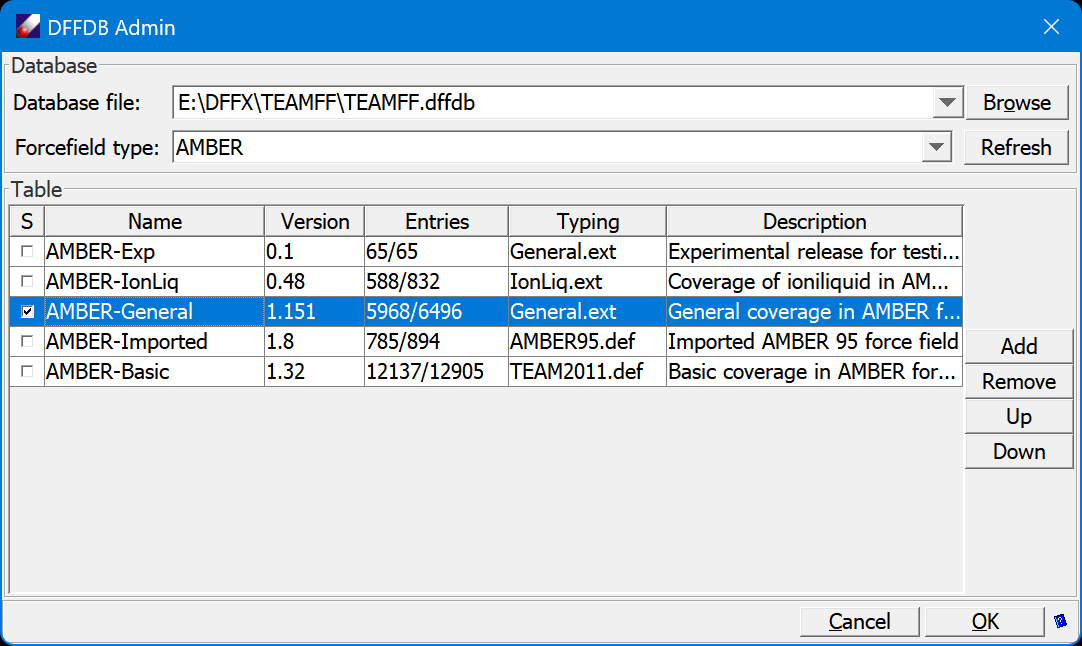
TEAMFF
TEAM stands for Transferable, Extensible, Accurate and Modular. It represents a novel force field, in which the parameters are associated with molecular fragments and these fragments and their parameters are stored in computer databases. Consequently, the force fields are more transferable and accurate than previously existing ones. Due to the database technology, they are unlimitedly (other than the hardware limitation) extensible. To use the TEAM force field is the same as to use any other ��normal�� force fields. You input a model (liquid, polymers, protein/ligand complexes) to the software, the software automatically identify the fragments required, finds the related force field parameters, and composes a force field for the model. Meanwhile, it assigns the atom types and atomic charges for the model. The result model and force field parameters can be used subsequently by the simulation engines directly. The TEAM force field databases are made in different versions based on applications and force field types. Currently released TEAM force fields are for material science applications �C they contain parameters for most over 500 polymers and many common organic molecules, and for the following force field types: AMBRR, CHARMM, CFF and GROMACS.
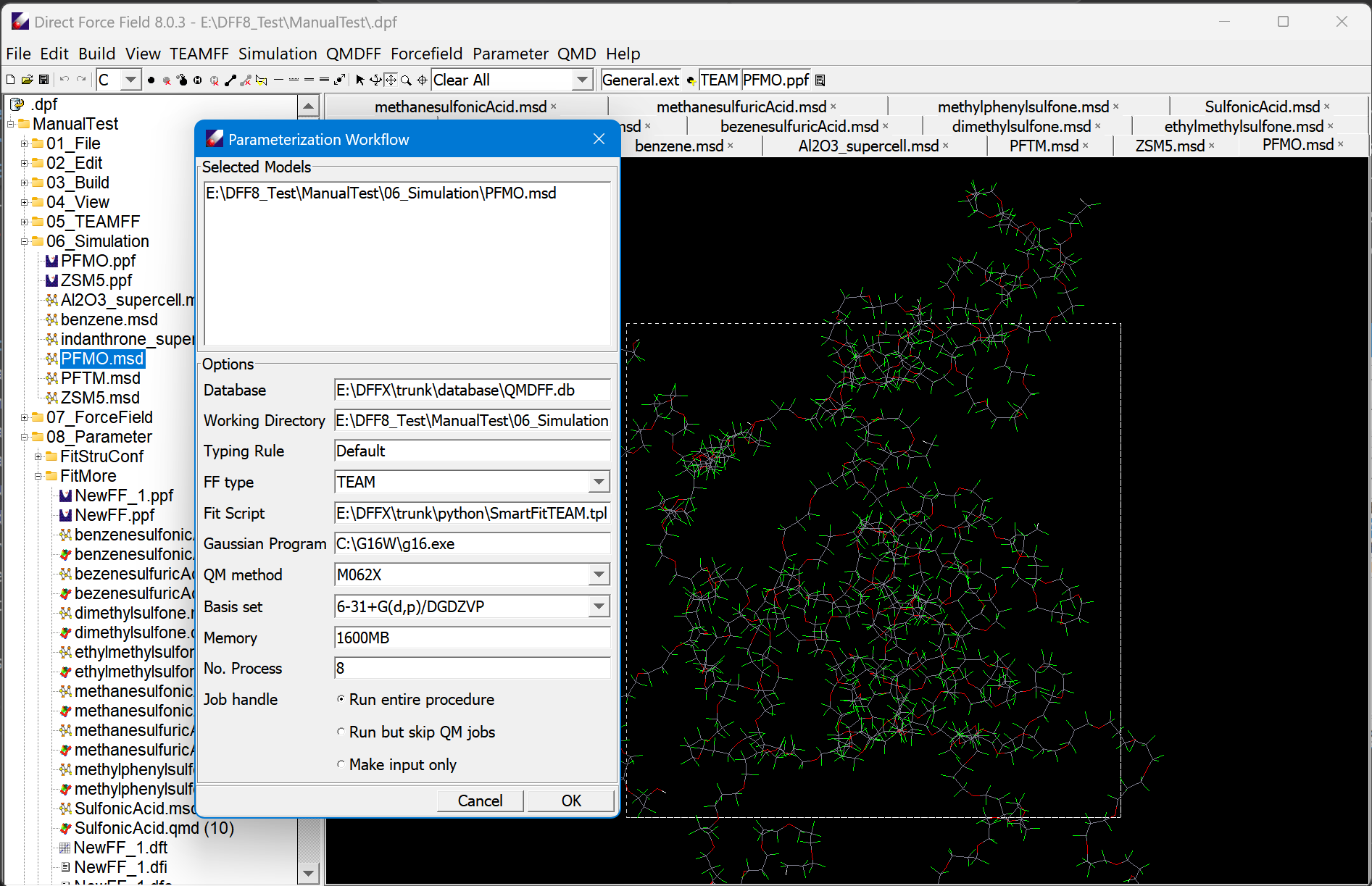
Automatic parametrization workflow
Automatic Parameter Generator (APG) functions based on the fitting engine (FIT), the database manager (DBM) and the Automatic Data Generator (ADG). A patent-pending technique fully automates the entire parameterization process. From a target molecule, it identifies new (unknown) fragments in the selected TEAM databases, generates sample configurations, performs required ab initio calculations, generates force field parameters using the ab initio data, validates the fitting results, and stores the results into the TEAM databases. All of these used to be very time-consuming processes can be performed as easy as a one-button-touch now.